【病例报告】表现为反复高热和网状青斑及脑梗死的腺苷脱氨酶2缺乏症患者一例
时间:2024-10-02 06:04:09 热度:37.1℃ 作者:网络
摘要:腺苷脱氨酶2缺乏症(DADA2)是近年来发现的一种罕见的血管炎相关的单基因、常染色体隐性遗传病。作者报道了1例成年确诊的DADA2病例,患者在儿童期出现反复高热伴网状青斑,后多次发生脑梗死,遗留明显运动功能障碍、四肢肌张力增高、双足内翻畸形和步态异常。该患者经基因和血清腺苷脱氨酶检测确诊,应用抗肿瘤坏死因子治疗后1年未再复发卒中,经综合康复训练肌张力下降,异常步态改善,日常生活活动能力提高。通过报道该例病例,以期为神经内科医师在临床工作中提供部分借鉴。
腺苷脱氨酶2缺乏症(deficiency of adenosine deaminase 2,DADA2)是近年来发现的一种罕见的血管炎相关的单基因、常染色体隐性遗传病,该病为腺苷脱氨酶2(adenosine deaminase 2, ADA2)基因纯合或复合杂合变异导致ADA2活性低下或缺乏所致,表现为自身炎症反应综合征、卒中反复发作、类似结节性动脉炎的血管炎及免疫缺陷等一组症候群。该病早期诊断困难,笔者报道1例成年后方确诊的DADA2的病例,以期为神经内科医师在临床工作中对该病的诊疗提供部分借鉴。
患者
男,23岁,因“反复高热伴皮疹18年,行走不稳、口角歪斜15年,肢体僵硬无力7年”于2023年4月13日入住中国康复研究中心(北京博爱医院)神经内科治疗。患者18年前(5岁)开始出现反复高热,体温波动在38~40℃之间,伴有全身不规则网状青斑,解热退烧药物无效,口服泼尼松片可降温(具体剂量不详),体温正常后停用糖皮质激素药物。此后每年反复发作发热4~6次,四肢网状青斑遇冷出现,遇热消退。患者15年前(8岁)行走时突发走路不稳跌倒,就诊于外院,头部MRI示“脑桥异常信号”,诉腰椎穿刺脑脊液压力及常规生化等检查未见异常(具体不详),10余天后所有症状均完全缓解,诊断“癫痫?”。患者7年前(16岁)逐渐出现肢体僵硬,以左上肢和双下肢明显,构音不清,就诊于当地医院,头部MRI示“桥脑、双侧丘脑、基底节可见多发、点片状长T1及长T2信号,部分高弥散信号”,未明确诊断,给予对症营养神经治疗后症状无改善。5年前就诊于外院,诊断“脑小血管病,小血管炎不除外”,右上肢皮肤活检提示“血管炎性改变”,诊断“恶性萎缩性丘疹病(Degos病)”,先后2次给予静脉丙种球蛋白治疗(0.4g/kg,连续使用5d,间隔日期不详),症状略有改善。6月余前突发左侧肢体力弱,于当地医院住院给予丙种球蛋白、阿司匹林0.1g/d、阿托伐他汀20mg/d治疗,具体诊治经过不详,症状无改善,后就诊于外院,实验室检查结果示血腺苷脱氨酶降低(2.3U/L;参考范围:4.0~24.0U/L),ADA2明显减低(0.1U/L;参考范围:2.0~12.0U/L),基因检测结果示“ADA2基因纯合变异:C.139G>C(P.gly47Arg)”诊断为“DADA2”,给予阿达木单抗40mg,2周1次,抗肿瘤坏死因子治疗,加用乙哌立松5mg(3次/d)、巴氯芬10mg(3次/d)对症降低肌张力治疗,肢体僵硬及步态异常等症状无明显改善。既往史:患者3年前外伤性蛛网膜下腔出血,外院保守治疗未遗留后遗症。吸烟史6年余,偶饮酒。家族史:母亲体健,父亲5年前逐渐出现尿失禁、四肢及躯干僵硬,外院诊断“多系统萎缩”,发病4年后去世,具体不详。否认父母近亲婚姻。有一姐姐,体健。入院体格检查:体型瘦小,身高163cm,体质量54kg,体质量指数20.32kg/m2,四肢远端皮肤可见网状青斑(图1)。神经系统体格检查:意识清楚,精神可,语速略缓慢,理解力、记忆力、定向力、计算力(100-7-7-7-7=?)下降。双侧瞳孔等大等圆,直径3mm,直接及间接对光反射灵敏,双眼视力下降,1米以远视物模糊,其余脑神经检查未见异常。肌力左上肢近端Ⅴ-级,远端Ⅳ级,左下肢Ⅴ-级,右侧Ⅴ级,左上肢及双下肢肌张力增高,屈肌及伸肌张力均增高,双上肢屈肌为主,双下肢伸肌为主,被动活动伴有关节和肌肉疼痛。四肢腱反射亢进,双侧髌阵挛、踝阵挛阳性,双侧掌颌反射、噘嘴反射、双侧Hoffman征、Babinski征阳性,双侧指鼻试验、跟膝胫反射缓但稳准。深浅感觉正常对称。起身缓慢费力,站立后身体前倾,脊柱右侧侧弯,左足内翻,痉挛步态。Holden步行能力2级,日常生活活动能力评分60分。实验室检查:CD3T细胞:733个/μl↓(参考范围:940~2140个/μl),CD4T细胞:345个/μl↓(参考范围:550~1200个/μl),CD8T细胞:303个/μl↓(参考范围:380~790个/μl)。白细胞介素(IL)6∶39.1ng/L↑(参考范围:<5.9ng/L),IL-8∶228ng/L↑(参考范围:<62ng/L),ADA2:0.1U/L↓(参考范围:2.0~12.0U/L),腺苷脱氨酶:2.3U/L↓。患者母亲ADA2:1.8U/L↓,腺苷脱氨酶:5.2U/L↓。患者头部MRI(平扫+磁敏感加权成像)示双丘脑、基底节多发点片状T1长信号、T2异常信号,T2液体衰减反转恢复(FLAIR)序列呈高信号,多发微出血灶(图2)。二代测序技术-全外显子基因检测受检者在Sneddon综合征、血管炎-自发性炎症-免疫缺陷和血液学缺陷综合征相关基因ADA2存在一处纯合变异:c.139G>C(p.Gly47Arg),为可以解释患者表型的致病性变异(图3)。结合患者临床症状、体征(全身炎症、网状青斑和早发性卒中)、血腺苷脱氨酶检测、头部MR检查和ADA2基因检测结果,疾病诊断:DADA2。药物治疗:继续给予阿达木单抗40mg(2周1次)治疗,逐步调整降肌张力药物为替扎尼定1mg(2次/d)、盐酸乙哌立松25mg(3次/d)及巴氯芬10mg(2次/d)。康复诊断:肢体运动功能障碍,肌张力障碍,关节活动受限,平衡障碍,步行障碍,日常生活活动能力依赖。康复治疗:日常佩戴双踝足矫形器联合智能行走机器人训练矫正足内翻畸形、辅助站立和步行,通过物理治疗、作业治疗手法改善肌张力障碍,给予文娱疗法、动能医师情景互动训练系统改善患者动作协调性和躯干平衡能力,给与温热水疗辅助降低肢体及躯干肌张力、缓解痉挛造成的疼痛。患者共住院康复治疗30d,出院6个月后电话随访无卒中复发,肌张力明显下降,日常佩戴矫形器可独立步行(Holden步行能力3级),日常生活能力提高(日常生活活动能力评分75分)。
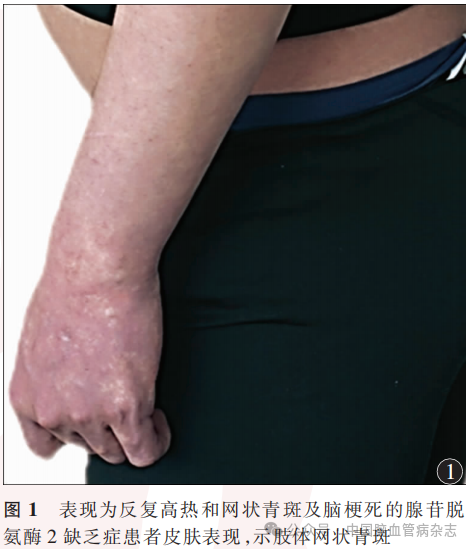
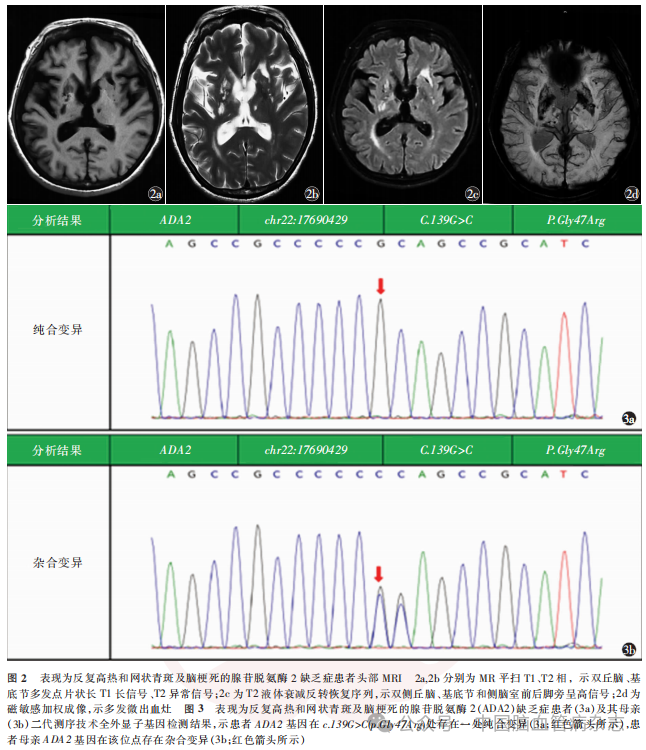
讨论
ADA2是主要的细胞外腺苷脱氨酶,在临床中可通过分光光度法和氨试剂法测定其在血清或血浆中的含量,另外还可以通过酶联免疫吸附试验或蛋白免疫印迹来定量,后两者临床不常用。ADA2数量减少和功能缺失可导致全身血管炎性病变。DADA2是一种罕见的ADA2单基因隐性遗传性自身炎症性疾病,该病最早在2014年被发现,以全身性炎症、早发性卒中和免疫缺陷为特征。一项基于文献报告的研究纳入2018年7月1日前发表文献中的160例DADA2患者,结果显示,此类患者的皮肤受累最常见,约占90%,其中网状青斑占72.0%(90/125)、皮肤血管炎占57.6%(72/125),其他症状包括发热[63.3%(69/109)]、卒中[38.0%(57/150)]、神经病变[42.6%(43/101)]、低免疫球蛋白M56.7%(51/90)]、低免疫球蛋白G[52.5%(53/101)]、白细胞减少[61.1%(55/90)]、贫血[53.9%(55/102)]和血小板减少[32.5%(25/77)]。DADA2患者最初可表现为慢性、轻微的症状,如皮肤损害,也可能出现某些急性严重的表现,如卒中(包括缺血性卒中和出血性卒中);该病的患者临床表现差异较大,即使同一纯合子变异体患者(即使同一家族)之间在发病年龄、临床症状和严重性方面也可能有很大不同,可能与遗传修饰作用、残存ADA2活性数量和环境因素有关。单基因的突变如何转化为临床表现的广泛性和疾病的异质性仍有待于进一步探索。
目前全球已报道的DADA2确诊病例有600多例(https://dada2.org/),在遗传学上,有害的ADA2变异体的总携带率约为1/236,全世界预估有35000人患该病。77%DADA2患者在10岁前确诊,30岁之前的病死率为8%(https://dada2.org/)。目前遗传性自身炎性疾病突变登记网站报道的ADA2致病突变位点和可能致病突变位点有159处(https://infevers.Umai-montpellier. fr/web/index.php)。理论上必须在两条等位染色体上同时出现致病性变异才有可能致病(纯合或复合杂合变异致病),最常见的基因变异位点为P.Gly47Arg(P.G47R)、P.Gly47Ala (P.G47A)、P.Arg169Gln (P.R169Q)和P.Tyr453Cys (P.Y453C)。本例患者ADA2基因存在一处纯合变异:c.139G>C(p.Gly47Arg),该变异位点已报道为致病变异。家系验证结果表明,其母亲此位点存在杂合变异。患者母亲否认近亲结婚。遗憾的是本例患者父亲已过世,无法进行ADA2基因的测序比对。
DADA2患者可出现缺血性卒中和出血性卒中,本例患者的头部MRI发现缺血和出血性改变。研究表明,DADA2患者缺血的表现以脑小血管闭塞导致腔隙性梗死多见,头部磁共振可表现为急、慢性小面积皮质下梗死,累及脑深部核团和脑干,同时可发现深部的微出血灶。
DADA2可导致广泛的血液学异常和免疫缺陷,临床中对儿童时期全身炎症起病、伴有皮肤改变等可疑临床特征的患者应进行基因筛查和(或)ADA2酶活性检测,以建立早期诊断,早期诊断DADA2是减少患者全身器官损害,改善预后的关键。本例患者儿童期起病,反复高热,多次脑梗死伴有网状青斑,血ADA2减低,头部MRI多发埂塞灶和微出血灶,ADA2基因存在纯和变异。该病一经确诊,应尽早治疗。根据临床经验和2023年发表的DADA2国际共识,目前肿瘤坏死因子抑制剂(tumor necrosis factor inhibitor, TNFI)是治疗DADA2的主要药物,可以减轻自身炎症,缓解免疫缺陷、肝脾肿大和中性粒细胞减少,改善生长发育、贫血和血小板减少,显著降低缺血性和出血性卒中及其他脏器血管炎的发生风险,该类药物包括依那西普、阿达木单抗、戈利木单抗、英夫利昔单抗和赛妥珠单抗。对于确诊DADA2且有临床症状的患者,TNFI应立即且长期应用,直至更有效的药物出现。多项研究显示,糖皮质激素、抗风湿药物、免疫抑制剂、IL-1和IL-6的生物靶向药物治疗DADA2效果欠佳。另外,因该病具有潜在的脑出血风险,不建议使用抗血小板聚集药物或抗凝血药物。对于有骨髓衰竭、难治性免疫缺陷的DADA2患者,可考虑造血干细胞移植。一项多中心研究纳入了6个国家共14例DADA2患者,均进行造血干细胞移植治疗,平均随访18个月,结果显示,患者血液及免疫学指标均控制良好。重组ADA2蛋白和基因治疗目前尚处于研究阶段。2023年DADA2国际共识推荐DADA2患者应每3~6个月门诊随访1次,进行体格检查、常规实验室检查,根据患者的病情必要时可行相应的影像学检查。
目前尚无该病相关康复治疗的报道。本研究报道的DADA2患者发病至确诊的时间较长,在卒中反复发作后遗留有明显运动功能障碍,包括肌力下降、肌张力障碍、关节挛缩、步态异常,且伴有疼痛,以致日常生活活动能力明显下降,无法正常工作。在联合1个月的康复治疗后,患者Holden步行能力提高至3级(患者需要1人口头管理或伴行而无身体上接触),日常生活活动能力评分提高至75分(轻度功能缺陷)。因此,我们建议对于已发生卒中且遗留有功能障碍的DADA2患者,在药物治疗同时,应至专业的康复医院进行康复治疗,必要时可佩戴矫形器具,以期有最大程度的获益。

